Lyme-Associated Parkinsonism: A Neuropathologic Case Study and Review of the Literature
David S. Cassarino, MD, PhD,a Martha M. Quezado, MD,a Nitya R. Ghatak, MD,a and Paul H. Duray, MDa
|
aFrom the Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, Md (Drs Cassarino, Quezado, and Duray); and the Departments of Pathology and Neuropathology, Virginia Commonwealth University, Richmond (Dr Ghatak) |
Accepted April 11, 2003
Neurological complications of Lyme disease include meningitis, encephalitis, dementia, and, rarely, parkinsonism. We present a case of striatonigral degeneration, a form of multiple system atrophy, in Lyme-associated parkinsonism. A 63-year-old man presented with erythema migrans rash, joint pains, and tremors. Serum and cerebrospinal fluid antibodies and polymerase chain reaction for Borrelia burgdorferi were positive. Clinical parkinsonism was diagnosed by several neurologists. Despite treatment, the patient continued to decline, with progressive disability, cognitive dysfunction, rigidity, and pulmonary failure. At autopsy, the brain showed mild basal ganglia atrophy and substantia nigra depigmentation, with extensive striatal and substantia nigral neuronal loss and astrogliosis. No Lewy bodies were identified; however, ubiquitin-positive glial cytoplasmic inclusions were identified in striatal and nigral oligodendroglia. There were no perivascular or meningeal infiltrates, the classic findings of neuroborreliosis. To our knowledge, this is the first report of striatonigral degeneration in a patient with B burgdorferi infection of the central nervous system and clinical Lyme-associated parkinsonism.Lyme disease is an infection caused by Borrelia burgdorferi, a spirochete transmitted by Ixodes ticks in the United States. Patients often initially present with the classic erythema migrans rash, a macular, erythematous, circular area with central clearing that expands around the site of the tick bite. The rash usually begins within 3 to 30 days after the bite, but is only found in about 60% of patients.1 Patients with long-standing Lyme disease may develop myocarditis, oligoarthritis of large joints, and central nervous system involvement (typically meningitis, encephalitis, and cranial neuropathy, and, rarely, basal ganglia and cognitive dysfunction) in the tertiary phase of the disease. There have also been reported cases of patients with Lyme disease developing clinical parkinsonism.2–6 We describe what to our knowledge is the first such case with autopsy follow-up.
Patients with Lyme meningitis usually show increased numbers of lymphocytes and plasma cells in the pia and arachnoid, with some atypical lymphocytes.1 In Lyme encephalitis, there is edema, microglial activation, and intraparenchymal lymphoplasmacytic infiltrates in a predominantly perivascular distribution.1 These findings were lacking in the current case. Instead, the brain showed neuronal loss, gliosis, and glial cytoplasmic inclusions in the striatum and substantia nigra, leading to the diagnosis of striatonigral degeneration (SND).
Striatonigral degeneration is now recognized to be a subtype of multiple system atrophy (MSA), a relatively uncommon neurodegenerative disease characterized by neuronal loss and astrocytosis of the basal ganglia and substantia nigra, with characteristic ubiquitin-positive glial cytoplasmic inclusions.7 These inclusions contain -synuclein, which can be identified immunohistochemically in glial cells. To our knowledge, the presence of glial cytoplasmic inclusions and synuclein has not been previously reported in the brains of patients with Lyme disease.
The patient was a previously healthy, 63-year-old white man who presented with an erythema migrans rash on his left inner thigh in June 1995. He developed diffuse musculoskeletal pain, swelling of the left knee, tremor of the left hand, and pain in the left shoulder and arm during the subsequent year. In June 1996, the diagnosis of Lyme disease was made based on a serum Western blot showing B burgdorferi–specific immunoglobulin (Ig) G bands. The patient's musculoskeletal pains and hand tremor worsened during the next few months, with loss of function. He was treated with 3 weeks of intravenous (IV) ceftriaxone without improvement in August 1996. A magnetic resonance imaging scan of the head and neck was reportedly normal in February 1997. He resumed antibiotic therapy with 2 weeks of IV ceftriaxone and then 42 days of IV cefotaxime sodium, with little improvement in his condition. In May 1997, a neurology consult was obtained, at which time a spinal tap cerebrospinal fluid (CSF) was found to be positive by enzyme-linked immunosorbent assay (ELISA) for B burgdorferi-specific IgG. Neurological examination documented parkinsonism, which was attributed to Lyme neuroborreliosis. Pharmacological treatment was initiated, without apparent benefit.
By July 1998, the patient had lost 20 kg and had developed symptoms, including chronic fatigue, tremors, and neck and bilateral hand pain; his movements were stiff and painful. He also developed cogwheel rigidity in August 1998. Due to continued clinical deterioration, he was started on oral antibiotics, including clarithromycin, ciprofloxacin, and hydroxychloroquine. His tremors seemed to improve after treatment; however, his other symptoms continued unabated. Western blots for B burgdorferi–specific IgM (30, 34, 41, and 93 kd) and IgG (30, 39, 41, 58, 66, and 93 kd) antibodies were positive in November 1999. Despite continued antibiotic treatments, the patient's movement disorder continued to progress. By May 2000, he exhibited decreased memory, incontinence, drooling, and inability to ambulate independently or to care for himself. Cerebrospinal fluid and blood polymerase chain reaction (PCR) tests at that time for Borrelia species were positive, and PCR for Babesia species was negative. A red blood cell culture showed classic spirochetes in his red cells. Oral multiagent antibiotic therapy was continued.
In December 2000, the patient was admitted to the hospital for aspiration pneumonia and was treated with antibiotics and parenteral nutrition. He was readmitted in January 2001 for another episode of aspiration pneumonia. He had a sputum culture that was positive for Staphylococcus aureus, and he was treated with IV vancomycin. In February 2001, a sputum culture was reportedly positive for B burgdorferi. A repeat serum Western blot for Borrelia IgM and IgG was positive, and PCR for Babesia organisms was also positive. Despite continued antibiotic treatments (IV vancomycin, azithromycin, and atovaquone), the patient's neurological status continued to decline, and he finally succumbed to infection and respiratory failure in April 2001. A full autopsy was performed.
Gross examination revealed few significant findings. Externally, there were multiple bruises and IV marks, and decubital ulcers over the sacrum. The chest cavities contained fluid and there were bilateral pleural effusions. The brain and spinal cord were externally unremarkable; on sectioning, the basal ganglia showed mild atrophy bilaterally (Figure 1 ), with greater changes on the left, and the substantia nigra showed depigmentation. The cerebellum also appeared to show mild atrophy.
Microscopic examination of the heart showed scattered lymphocytes and plasma cells, with areas of mild fibrosis, suggesting possible remote myocarditis. No significant inflammation was identified in any other organs. The brain showed extensive neuronal loss and severe astrogliosis in the striatum (Figure 2 ) and substantia nigra (Figure 3 ). Other brain regions were unaffected. No Lewy bodies were identified; however, there were ubiquitin-positive glial cytoplasmic inclusions in scattered oligodendroglia in the striatum (Figure 4 ) and substantia nigra, but not in the pons (including the olivary nuclei) or cerebellum. These glial cytoplasmic inclusions also stained positively with synuclein immunohistochemistry (Figure 5 ). Premortem Western blot and ELISA studies showed positive reactions for Borrelia-specific IgM and IgG antibodies in both serum and CSF samples (Tables 1 and 2 ). Polymerase chain reaction analysis for Borrelia-specific sequences in the substantia nigra and basal ganglia was performed; however, the results were not able to be confirmed on the postmortem tissue.
To the best of our knowledge, this report describes the first case of parkinsonism arising in association with Lyme disease to come to autopsy. Histological study of the brain displayed characteristic morphologic changes of SND, a variant of MSA. The patient's diagnosis of Lyme disease was well documented, confirmed by both serum and CSF ELISA, Western blots, and premortem PCR studies. The patient developed signs and symptoms of MSA after his presentation with the erythema migrans rash, and there was no prior history of neurologic dysfunction. Although it cannot be excluded that the SND could have developed independent of his Lyme disease, the temporal association with tertiary Lyme disease, the high titer of Borrelia antibodies in his CSF, and premortem PCR for B burgdorferi-specific sequences in the CSF favor an association. The fact that the classic inflammatory changes associated with Lyme disease were absent may indicate an atypical central nervous system infection in this patient, or merely that the infection and inflammation had resolved by the time of death (which occurred 5 years after infection and after multiple courses of antibiotics). In most cases, the organisms cannot be identified in histologic sections.1,6
Regardless of whether the infection had resolved by the time of death, we hypothesize that it was sufficient to cause ongoing neuronal loss and astrogliosis leading to SND. Therefore, the negative studies for organisms in the postmortem tissue may reflect either the absence of organisms or the persistence of low numbers of spirochetes and false-negative findings. Overall, we believe that the SND and resulting parkinsonism in this case might be related to direct infection by Borrelia organisms, or to the immune response against the organisms, and these findings are therefore of particular interest because the etiology of SND and MSA is unknown.
Clinical diagnosis of MSA is based on diagnostic criteria, including parkinsonism with poor or transient response to L-dopa therapy. Patients often develop progressive bulbar dysfunction leading to dysphagia and laryngeal stridor, eventually predisposing to aspiration pneumonia.7 Our patient's parkinsonism was resistant to traditional medications, and he developed classic signs of parkinsonism as well as dysphagia, consistent with the clinical course of MSA. In a previous report of Lyme-associated extrapyramidal features in 5 patients,5 all of the patients exhibited akinesia, pains, and rigidity, similar to our patient, although only 2 developed tremors. Four of the 5 patients also developed bulbar dysfunction, a characteristic finding in MSA. Although none of these patients came to autopsy, and therefore could not be definitively diagnosed with MSA, the clinical findings were consistent with this conclusion and were generally similar to findings in our case. One significant difference was that their patients responded to anti–Parkinson's medications, which is unusual in MSA, and they also improved on antibiotics. This dissimilarity may indicate a different underlying pathology compared to the present case, in which there was little or no improvement with anti-Parkinson's drugs and antibiotics. Alternatively, as our patient did not receive antibiotics until 14 months after initial infection, he may have suffered irreversible neuronal damage by the time treatment was initiated.
Autopsy brain studies on patients with Lyme disease are limited to single case reports or small case series. In addition to meningoencephalitis, multiple other neuropathologic findings have been reported. One patient was found to have rhomboencephalitis on autopsy, with microgliosis and obliterative inflammatory vasculopathy associated with ischemic infarcts.2 Another case showed multifocal inflammation, neuronal cell loss, demyelination, and astrocytosis in the cortex, thalamus, cerebellum, and spinal cord.3 Bertrand et al4 reported 3 cases, 1 of which showed cortical involvement, and all 3 of which showed cerebral and cerebellar white matter changes, with associated lymphocytic infiltrates, microglial activation, spongiform changes, diffuse astrogliosis, and demyelination. To date, however, no neuropathologic findings have been reported in the substantia nigra or basal ganglia. Clinically, Kohlhepp et al5 described 5 patients with Lyme disease with extrapyramidal symptoms and documented CSF infection by B burgdorferi. Interestingly, treatment of the patients with high-dose penicillin led to both normalization of their CSF and improvement in their extrapyramidal symptoms.5
In primates infected with B burgdorferi, brain autopsy and PCR analysis showed organisms in the leptomeninges, nerve roots, and dorsal root ganglia, but not in the brainstem, cerebellum, or basal ganglia.6 Histologic and immunohistochemical studies with polyclonal anti–B burgdorferi antibodies confirmed the PCR results in this study.6
In summary, this is the first published report of SND or MSA, with characteristic ubiquitin and synuclein–positive inclusions, in a patient with documented B burgdorferi infection of the central nervous system and clinically diagnosed Lyme-associated parkinsonism. Therefore, this case raises the possibility of a causal link between B burgdorferi infection of the central nervous system and SND.
Acknowledgments
We thank Robert G. Beitman, MD, and Gregory P. Bach, DO, for submitting this fascinating case to us.
References
1. Duray, PH, and FW. Chandler. Lyme
disease. In: Connor DH, Chandler FW, Schwartz DA, Manz HJ, Lack EE, eds. Pathology
of Infectious Diseases. Stamford, Conn:
Appleton and Lange; 1997:635–646.
2. Kuntzer, T, J Bogousslavsky, and J
Miklossy. et al. Borrelia rhombencephalomyelopathy. Arch Neurol 1991;48:832–836. [PubMed Citation]
3. Kobayashi, K, C Mizukoshi, and T
Aoki. et al. Borrelia burgdorferi-seropositive chronic encephalomyelopathy:
Lyme neuroborreliosis?. An autopsied report. Dement Geriatr Cogn
Disord 1997;8:384–390.
4. Bertrand, E, GM Szpak, and E Pilkowska.
et al. Central nervous system infection caused by Borrelia burgdorferi:
clinico-pathological correlation of three post-mortem cases. Folia
Neuropathol 1999;37:43–51.
5. Kohlhepp, W, W Kuhn, and H.
Kruger. Extrapyramidal features in Lyme borreliosis. Eur Neurol 1989;29:150–155. [PubMed Citation]
6. Cadavid, D, T O'Neill, H Schaefer,
and AR. Pachner. Localization of Borrelia burgdorferi in the nervous system and
other organs in a nonhuman primate model of Lyme disease. Lab Invest 2000;80:1043–1054. [PubMed Citation]
7. Lowe, JS, and N. Leigh. In: Graham
DI, Lantos PL, eds. Greenfield's Neuropathology. New York, NY: Oxford University Press; 2002:343–346.
Table 1. Western Blot Results

Table 2. Enzyme-Linked Immunosorbent Assay
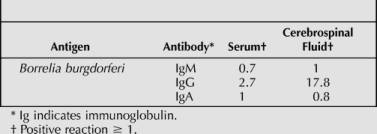
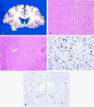
Figure 1. Coronal section of the brain showing basal ganglia atrophy.
Figure 2. Striatum with neuronal loss and gliosis (hematoxylin-eosin, original magnification ×100).
Figure 3. Substantia nigra with neuronal loss and gliosis (hematoxylin-eosin, original magnification ×100).
Figure 4. Ubiquitin-positive glial cytoplasmic inclusions in the striatum (hematoxylin-eosin, original magnification ×400).
Figure 5. Synuclein–positive glial cytoplasmic inclusions in the striatum (hematoxylin-eosin, original magnification *200)